Likely to be as important as oncogenes in cancer pathogenesis. It has been a long standing hope that despite the biological heterogeneity of cancer cells there would be common denominators for understanding, treating and preventing these diseases. The tumour suppressor proteins may play this role.
p53 acts in late G1 phase. It prevents the cell progressing to the S phase i.e. is antiproliferative. p53 function almost always disrupted in high grade tumour cells. The protein contains an N-terminal acidic transactivator domain, this in combination with the nuclear localization of p53 suggests a role in transcriptional control. Much direct evidence to support this conclusion. p53 is a tetrameric protein, it binds specifically to promoter element and specifically transactivates gene expression.
![]()
p53 mutations are common in many tumour cells, causing the protein to tend to lose its DNA binding and transactivation properties. Mutations also cause a conformational change in the protein (detected by using antibodies). This conformational change is transdominant and can be transmitted to "conformationally normal p53". Mixed oligomers lose their ability to transactivate transcription.
The normal conformation is antiproliferative. In addition to these intragenic mutations I have described numerous instances where viral proteins directly perturb p53 function by sequestering it or in one case by degrading it i.e the E6 protein of "high risk papilloma viruses such as HPV18. Can also be disrupted by a cellular protein made by the MDM2 gene, identified by virtue of its amplification in spontaneously transformed cell lines, product binds to p53 and prevents transactivation. Not an experimental curiosity this gene is amplified in a majority of human sarcomas. A simple model for p53 function proposed in the figure transactivates genes involved in down regulating cell growth or invasion. Expression is lost in a number of ways, loss of one or both alleles so p53 concentration drops. Nonsense mutations so oligomerization domain at C-terminus is lost or missense transdominant mutations. missense mutation in one allele usually results in the loss of the other allele through mitotic recombination resulting in the absence of any wt protein e.g. colon, bladder, brain lung and liver.
p53 also modulates the function of WAF-1 a 21kd protein which inhibits G1 cyclins. These are a class of proteins which control the cell cycle in the G1 phase.
In mice or humans harbouring germline mutations in p53, development is normal. Suggests p53 not involved in the normal control of the cell cycle. Develop tumours soon after birth. Thought that it plays an import role in controlling growth in stressed cells. For example, both X-ray or drug treatment of cells results in p53 expression and cell growth is arrested until the stress is removed and any DNA damage is repaired. So p53 is a molecular policeman which makes sure that the a cell is not stressed and that the DNA is in good shape. If it isn't p53 will not allow replication until the damage is repaired or it will trigger apoptosis. DNA tumour viruses have to knock out p53 function because they need the cells in S phase and this damage response must not be triggered. Tumour cells may progress through a stress phase e.g. anoxia or anueploidy and then inevitably select for cells with p53 disfunction.
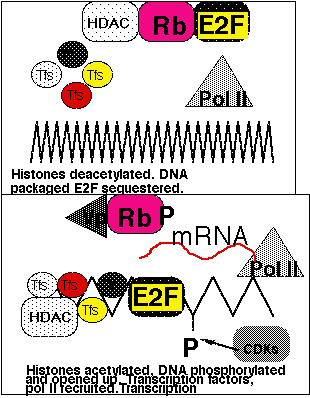
p53 exits in two allelic forms, Arginine R72 and Proline P72.These allelic variants are thought to be functionally equivalent and do not seem to be a factor in non viral induced tumourigenesis. However in 1998 the R72 variant was shown to be more susceptible to papilloma HPV18 E6 mediated proteolysis than the P72 form, both in vitro and in vivo. Furthermore cervical carcinomas were 7x more likely than expected to be expressing only the arginine allele.Possibly indicating that persons homozygous for Arg72 are more likely to develop cervical carcinoma following infection with HPV. This latter observation is currently contentious.RB1 is thought to act as a brake on the advancement of cells from G0/G1 into S phase. It is another target for all of the DNA viruses which has to be taken out if replication is to occur. Both copies of the gene need to be lost for oncogenesis, found in retinoblastomas, sarcomas, breast and bladder cancers etc. Best characterised cellular protein interacting with Rb is E2F, this is a transactivator. E2F activates proteins used in S phase such as TK and DNA pol alpha simplistic view is that Rb sequesters this protein in G1 and prevents transactivation. Mutation or phosphorylation results in loss of function. Hypophosphorylated in G1, hyperphosphorylated by cyclin dependent kinases at other times. Viral proteins bind to the hypo state preventing E2F sequestration suggesting that this is largely responsible for tumour suppression.E2F interacts with histone deacetylase. When the complex interacts with DNA histones are acetylated, DNA is phosphorylated and uncondenses and is available for transcription.
![]()
Not all tumour suppressor proteins act directly at the level of transcription. The tumour suppressor gene involved in neurofibromatosis (NF) encodes a protein which may exert negative control over ras. NF is a genetic disease characterised by tumours originating in the neural crest. Neurofibromin is a GTPase activating protein and converts active GTPras to inactive GDPras, effectors of ras not yet known. Not all tumour suppressor genes code for proteins.
Click the Back button to return to the lesson.